[ad_1]
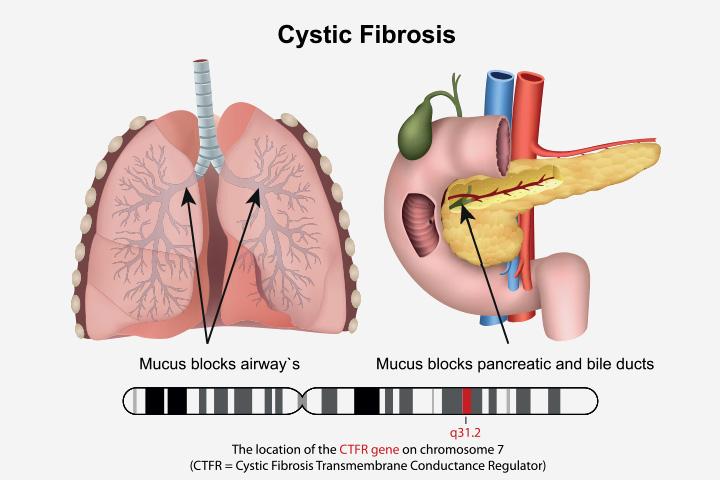
Image: Shutterstock
Cystic fibrosis is a progressive, genetic disease that causes sticky mucus to build up in the lungs and the digestive system. It may lead to frequent lung infections and may limit the breathing ability over time. It may also cause issues in digesting the food (1) (2).
The condition is genetically transmitted and is characterized by an abnormality in the body’s cells that regulate the production of salt, water, and mucus (3). It affects both males and females equally (4).
In this post, we tell you about the causes, signs, diagnoses, and treatment of cystic fibrosis in babies.
Causes Of Cystic Fibrosis In Babies
Cystic fibrosis is a genetic disorder. It happens when the gene that makes a protein responsible for the influx (going in) and outflux (coming out) of sodium and chloride in the cells is faulty. The faulty gene leads to the thickening of the mucus that lines the airways and other organs. It also makes the secretions of the mucous membranes stickier (5).
The thick and sticky mucus is difficult for the body to clear. It ultimately increases the risk of infections and interferes with the healthy functions of organs, causing various symptoms indicative of cystic fibrosis.
How Does A Baby Get Cystic Fibrosis?
A baby develops cystic fibrosis because they inherit a faulty gene from their parents. The gene that causes cystic fibrosis is recessive. Therefore, for a child to get cystic fibrosis, both the parents need to be carriers of the faulty gene.If only one parent is a carrier, the child won’t get the disease but still may be a carrier (4).
If both the mother and the father carry one copy of the cystic fibrosis gene, but they do not have the disease themselves, their children may have a (6):
- 25 percent chance of getting both defective copies and developing cystic fibrosis
- 50 percent chance of inheriting one defective copy and being only a carrier
- 25 percent chance of neither being a carrier nor having the disease
Approximately 30,000 people in the United States have cystic fibrosis. It is estimated that about 10 million people are carriers of the cystic fibrosis gene (4).
Signs And Symptoms Of Cystic Fibrosis In Babies
The signs and symptoms of cystic fibrosis usually begin to show within the first year of life. It affects different organs and systems, leading to various symptoms. General signs and symptoms could be evident to the parents, while most organ-specific symptoms are detectable during a medical examination.
General symptoms of cystic fibrosis (7):
- Slow growth
- Less weight gain
- No stool in the first 24 to 48 hours of life
- Skin with a salty taste
Respiratory symptoms of cystic fibrosis (6):
- Chronic cough
- More mucus in lungs and sinuses
- Tiredness
- Frequent lung infections
- Frequent sinus infections
- Coughing up blood
- Heart enlargement
- Nasal polyps
- Sinusitis
- Symptoms of pneumonia (fever, increased coughing, shortness of breath, increased mucus, and loss of appetite)
Digestive symptoms of cystic fibrosis (6):
- Belly pain from constipation
- Increased gas, bloating, or a distended belly
- Loss of appetite
- Nausea
- Pale or clay-colored, foul-smelling, and mucus-rich stool that floats
- Blocked pancreatic ducts
- Problems with the absorption of key nutrients, such as fats, proteins, and fat-soluble vitamins like A, D, E, and K, causing malnutrition
Diagnosis Of Cystic Fibrosis In Babies
Newborns are usually screened for cystic fibrosis. Most babies are diagnosed before the age of two years. In rare cases, the disease is not detected until the age of 18 years (7).
A blood sample is collected from the baby’s heel in the first few days of life. After the initial screening, if needed, the doctor tests the baby’s sweat to determine its sodium and chloride content. A high level of salt (sodium and chloride) indicates cystic fibrosis (5) (8).
In most cases, cystic fibrosis is diagnosed in the initial screening process. However, in some babies, it is diagnosed following some other unexplained illness. In these babies, either a sweat test (as babies with cystic fibrosis have more salt in their sweat) or a genetic test is done. Genetic testing is done by collecting blood samples or an inner cheek swab (5).
Management Of Cystic Fibrosis
There is no cure for cystic fibrosis. The treatment plan depends on the symptoms’ severity and could change as per the baby’s age. The objective of the treatment is to reduce the intensity of symptoms, mitigate organ issues, and facilitate healthy development.
A treatment plan for cystic fibrosis could consist of the following measures (9) (10).
- High-calorie diet: Some babies with cystic fibrosis need a high-calorie diet for healthy growth and development.
- Pancreatic enzymes: Babies with cystic fibrosis-related digestion issues may need to consume pancreatic enzymes before their meals. These pancreatic enzymes help digest the food, helping the baby attain optimum weight and growth.
- Vitamin supplementation: The disorder interferes with the absorption of various vitamins. Therefore, the doctor may prescribe vitamin supplements to ensure adequate nourishment.
- Mucus thinners: Medicines, such as dornasealfa (Pulmozyme), help thin the mucus. Thin mucus is easier to cough and expel.
- Bronchodilators: Medicines, such as albuterol, help open the airways, aiding the lungs in clearing the mucus.
- Antibiotics: They are used to treat bacterial infections if any.
- Hypertonic saline: Inhaling hypertonic saline mist draws more water into the airways. The water helps in thinning the mucus.
- Airway clearance therapy (ACT): This therapy loosens the sticky mucus to make it easier to clear it through coughing. It will help the baby breathe easier. The doctor will decide the best option from the various techniques and treatments available under airway clearance therapy.
Frequently Asked Questions
1. What is the life expectancy of a baby with cystic fibrosis?
The average life expectancy of people affected by cystic fibrosis is around 44 years (7).
2. Do babies with cystic fibrosis poop a lot?
Yes, it is possible for some babies with cystic fibrosis to pass more stools than usual (11).
Cystic fibrosis is an irreversible condition and requires lifelong care. There is no way to prevent the condition. If you have or your partner has a family history of cystic fibrosis, you may consider genetic testing before becoming pregnant. You may also ask your doctor to perform a screening test on your newborn if it is not already included in your baby’s screening tests list.
References:
[ad_2]
Source link




: Symptoms, Treatment & More")

{kind=link}